标题: 科学家发现两个恢复皮肤关键胶原蛋白含量的药物 [打印本页]
作者: cooldaddy 时间: 2019-4-6 06:50
标题: 科学家发现两个恢复皮肤关键胶原蛋白含量的药物
不知什么时候,海参、猪蹄等等富含胶原蛋白的食物,被打上了美容养颜的标签。就连桃胶这种多糖组成的胶冻状食品,都要蹭一下胶原蛋白的热点,号称自己也有美容的功能。至于几乎是纯的胶原蛋白的阿胶,更是被炒上了天价。
虽说打着胶原旗号的美容食品基本都是收智商税的,但胶原美容这件事倒是真的。只不过这个补充方式嘛,倒不是说吃下去的蛋白质就绝对不能原样吸收,但这个吸收效率,看看屡战屡败的口服胰岛素制剂就知道了[1]。
近日,东京医科齿科大学的Nan liu和Emi K. Nishimura等研究发现,各种损伤因素的作用下,皮肤中一种叫做COL17A1的胶原减少,导致了皮肤的衰老。而且他们还找到了能有效“补充”COL17A1胶原,让皮肤变得更为“年轻”的方法。相关研究发表在Nature上[2]。

桃之夭夭,灼灼其华。但要想靠桃胶来美容,也就只能指望安慰剂效应了
皮肤作为我们身体的第一道防线,替我们承受着外界的风吹日晒雨淋,而这些都会对皮肤造成损伤,加速皮肤的衰老[3]。不良饮食、糖尿病等内在因素也同样会加速皮肤的衰老[4]。
衰老的皮肤厚度变薄,失去弹性,出现色素沉着,受伤后的愈合能力也大不如前。特别是其中COL17A1等构成半桥粒的胶原的减少,让衰老的皮肤变得尤为脆弱[5]。
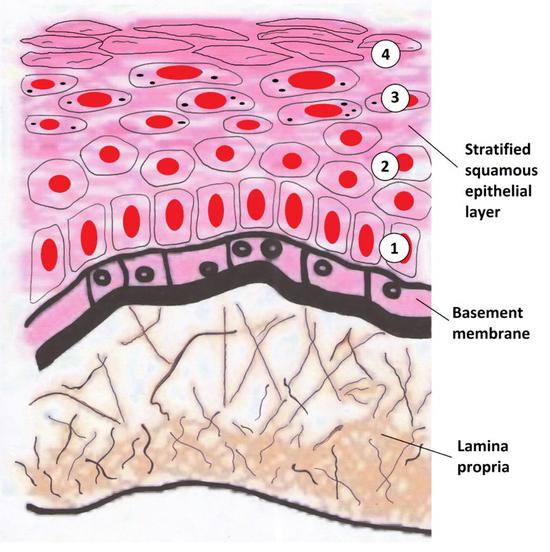
我们的皮肤包括了最外层由上皮组织构成的表皮,以及表皮内部结缔组织组成的真皮和皮下组织。在表皮和真皮之间,有一层薄薄的基底膜。表皮最深处的基底细胞,就是靠着半桥粒牢牢的附着在基底膜上[6]。而一碰就起血泡的大疱性表皮松解症,正是COL17A1等胶原的突变使得半桥粒失去了它的功能[7]。
除了把表皮附着在基底膜上,表皮最深处的这群基底细胞还是表皮其它细胞的来源,表皮的干细胞、祖细胞都在其中,对表皮的更新和损伤修复尤为重要。
为了进一步研究皮肤的衰老,研究人员在小鼠尾巴皮肤上进行了试验。与人类的皮肤一样,小鼠尾巴上的皮肤也分层良好,衰老时同样出现表皮细胞层数减少、基底细胞扁平化、细胞增殖活动减弱等现象。
除此以外,老龄小鼠尾部皮肤中,连接表皮和基底膜的半桥粒数量大幅下降。而对其中各种半桥粒组分的分析发现,与人类皮肤一样,小鼠尾部皮肤中胶原COL17A1含量随年龄增长而明显下降。
接下来,研究人员测试了紫外线、电离辐射、过氧化氢等各种加速皮肤衰老的理化因素对皮肤中COL17A1水平的影响,发现这些能造成基因组损伤的因素,都能让皮肤中COL17A1的水平下降。

敲除COL17A1使伤口愈合变慢,而过表达COL17A1促进了伤口愈合
而且,COL17A1缺陷的小鼠皮肤,更容易衰老,伤口愈合能力下降,受到紫外线照射时更容易出现色素沉着。而COL17A1的过表达,也让小鼠皮肤的伤口愈合能力更强,紫外线照射下更不容易出现色素沉着。
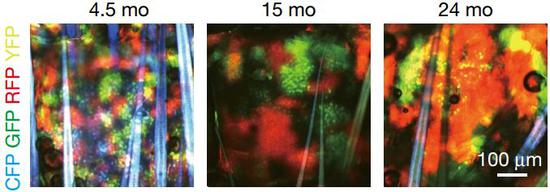
COL17A1的水平跟基因损伤有关,那它会不会影响细胞的增殖呢?研究人员让小鼠表皮细胞随机表达了红色、黄色、绿色或青色的荧光蛋白。在它们小时候,这些不同颜色的荧光细胞均匀地随机分布在皮肤上。但老了后,这些小鼠的皮肤中,出现了大块的单色区域。
更为重要的是,老年小鼠中,这些单色区域的大小,与COL17A1的表达水平正相关!
COL17A1高表达的细胞具有竞争优势!
进一步的研究发现,COL17A1阳性细胞倾向于进行水平分裂(也称对称分裂),生成两个同样连接在基底膜上的新细胞,占据更多的空间。而COL17A1阴性的细胞更爱进行垂直分裂(也称不对称分裂),其中一个子细胞进入表层的皮肤。再加上COL17A1阴性的细胞半桥粒功能不好,与基底膜连接不牢,很容易被周围COL17A1阳性的细胞挤出基底层。
这样的竞争,淘汰了那些COL17A1水平低的不健康表皮细胞,留下来COL17A1高表达的健康细胞,让我们的皮肤更为年轻。不过要是COL17A1高表达的细胞太少,产生的竞争压力有限,就不能有效淘汰那些COL17A1阴性的细胞了。
过表达COL17A1可以减缓皮肤衰老,但要想在人类皮肤中进行这一操作明显不太现实,研究人员转而去寻找那些能增加COL17A1表达的化合物。
最终,研究人员找到了两种化合物:Y27632和罗布麻宁。在体外,它们都可以增加COL17A1的表达,让表皮干细胞长出更多更大的克隆。而在体内,它们也都促进了伤口的愈合,起到了与过表达COL17A1相似的效果。
论文通讯作者Nishimura表示,这项研究最终可能会开发出面霜或口服药物,以阻止皮肤衰老,促进修复:“我们将与制药或化妆品公司合作,在临床使用这些药物。”
而接下来,他们还计划在其它上皮组织组成的器官中,研究类似的细胞间竞争在衰老中的作用。
| 欢迎光临 (http://ftp.zasq.net/~zazww/) |
Powered by Discuz! X3.2 |